Agonistas y antagonistas adrenérgicos
Acciones de catecolaminas y fármacos simpaticomiméticos
La mayoría de las acciones de las catecolaminas y los agentes simpaticomiméticos se pueden clasificar en siete grandes tipos:
- Una acción excitadora periférica en ciertos tipos de músculo liso, como aquellos en los vasos sanguíneos que irrigan la piel, los riñones y las membranas mucosas, y en células glandulares como las de las glándulas salivales y sudoríparas.
- Una acción inhibitoria periférica sobre otros ciertos tipos de músculo liso, como los que se encuentran en la pared del intestino, en el árbol bronquial y en los vasos sanguíneos que irrigan los músculos esqueléticos.
- Una acción excitadora cardiaca que aumenta la frecuencia cardiaca y la fuerza de contracción.
- Acciones metabólicas, tales como el aumento en la tasa de glucogenólisis en el hígado y los músculos y la liberación de ácidos grasos libres en el tejido adiposo.
- Acciones endocrinas, como la modulación (aumento o disminución) de la secreción de insulina, renina y hormonas hipofisarias.
- Acciones en el CNS, como estimulación respiratoria, aumento de la vigilia y la actividad psicomotora y reducción del apetito.
- Acciones presinápticas que tanto inhiben como facilitan la liberación de neurotransmisores, siendo la acción inhibidora la más importante fisiológicamente.
Las sustancias simpaticomiméticas, agonistas adrenérgicos o simplemente adrenérgicos, como su nombre lo indica, actúan como agonistas del sistema simpático simulando los efectos de las catecolaminas como la epinefrina, norepinefrina y dopamina. Pueden estimular directamente los receptores adrenérgicos o estimular la producción de noradrenalina en las terminaciones simpáticas. Estos fármacos elevan la presión sanguínea y tienden a ser bases debiles. La epinefrina es sintetizada por el cuerpo de la norepinefrina causando estimulación del SNC. Muchos de estos fármacos tienen acciones terapéuticas y pueden potencialmente causara abuso de sustancias, inducir tolerancia y provocar dependencia.
Las catecolaminas y los fármacos simpaticomiméticos se clasifican como simpaticomiméticos de acción directa, de acción indirecta o de acción mixta:
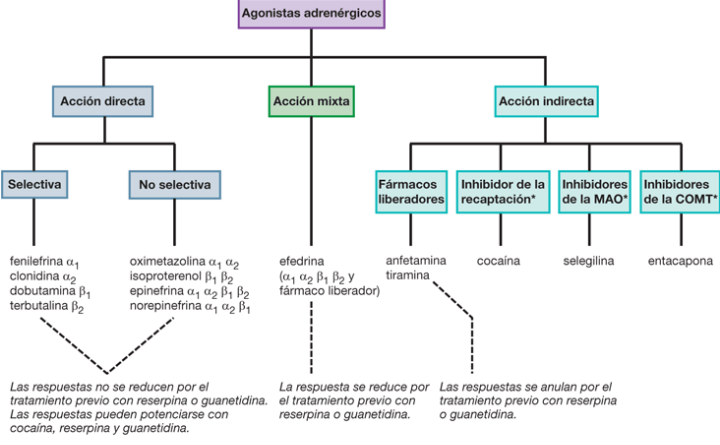
Los fármacos de acción indirecta aumentan la disponibilidad de la NE o la EPI para estimular los receptores adrenérgicos mediante varios mecanismos: 1) Al liberar o desplazar la NE de las várices nerviosas simpáticas, 2) al inhibir el transporte de NE a las neuronas simpática, lo que aumenta el tiempo de permanencia del transmisor en el receptor, 3) al bloquear las enzimas metabolizantes, MAO o COMT, aumenta con eficiencia el suministro del transmisor.
Los medicamentos que liberan NE de modo indirecto y también activan directamente los receptores se denominan fármacos simpaticomiméticos de acción mixta. Característica cardinal de estos medicamentos es que sus efectos se atenúan, pero no se eliminan, con el tratamiento previo con reserpina o guanetidina. Una característica de los fármacos simpaticomiméticos de acción directa es que sus respuestas no se reducen con el tratamiento previo con reserpina o guanetidina, que reducen la NE de las neuronas simpáticas. A causa de que las acciones de la NE son más pronunciadas en los receptores α y β1 que en los receptores β2, muchos fármacos no catecolamínicos que liberan NE tienen, predominantemente, efectos cardiacos y mediados por receptores α. Sin embargo, ciertos compuestos no catecolamínicos, con efectos tanto directos como indirectos sobre los receptores adrenérgicos.
La β-feniletilamina se puede ver como el compuesto original de las aminas simpaticomiméticas, que consiste en un anillo de benceno y una cadena lateral de etilamina. La estructura permite que se realicen sustituciones en el anillo aromático, los átomos de carbono α y β y el grupo amino terminal, para producir una variedad de compuestos con actividad simpaticomimética. Muchos fármacos simpaticomiméticos que actúan directamente influyen en los receptores α y β, pero la relación de actividades varía entre los medicamentos en un espectro continuo, desde una actividad con predominio α (fenilefrina) a una actividad con predominio β (INE). Las catecolaminas tienen un breve periodo de acción y son ineficaces cuando se administran por vía oral, porque se inactivan rápidamente en la mucosa intestinal y en el hígado, antes de llegar a la circulación sistémica. Los compuestos que no tienen uno o ambos sustituyentes hidroxilo no son activados por la COMT, y de este modo su eficacia oral y duración de acción se intensifican. Se han sustituido grupos distintos de hidroxilos en el anillo aromático. En general, la potencia de los receptores α se reduce y la actividad del receptor β es mínima; los compuestos pueden, incluso, bloquear los receptores β.
La sustitución de un grupo hidroxilo en el átomo de carbono β generalmente disminuye las acciones dentro del CNS, en gran parte porque reduce la solubilidad de los lípidos. Sin embargo, tal sustitución aumenta en gran medida la actividad agonista en los receptores adrenérgicos α y β. Aunque la efedrina es menos potente que la metanfetamina como estimulante central, es más potente para dilatar los bronquiolos y aumentar la presión sanguínea y la frecuencia cardiaca La sustitución de un grupo hidroxilo en el átomo de carbono β generalmente disminuye las acciones dentro del CNS, en gran parte porque reduce la solubilidad de los lípidos. Sin embargo, tal sustitución aumenta en gran medida la actividad agonista en los receptores adrenérgicos α y β. Aunque la efedrina es menos potente que la metanfetamina como estimulante central, es más potente para dilatar los bronquiolos y aumentar la presión sanguínea y la frecuencia cardiaca
Los factores importantes en la respuesta de cualquier célula u órgano a las aminas simpaticomiméticas son la densidad y la proporción relativa de los receptores adrenérgicos α y β. La respuesta final de un órgano blanco a las aminas simpaticomiméticas viene dictada no sólo por los efectos directos de dichos agentes, sino también por los ajustes homeostáticos reflejos del organismo. Uno de los efectos más llamativos de muchas aminas simpaticomiméticas es el aumento de la presión sanguínea arterial, provocado por la estimulación de receptores adrenérgicos α vasculares. El efecto del reflejo barorreceptor es de especial importancia para los medicamentos que tienen poca capacidad para activar los receptores β directamente. En enfermedades tales como la aterosclerosis, que pueden alterar los mecanismos barorreceptores, los efectos de los fármacos simpaticomiméticos pueden aumentar.
Catecolaminas endógenas
- Epinefrina
Es un potente estimulante de los receptores adrenérgicos α y β, y sus efectos sobre los órganos blanco son, por tanto, complejos. Su acción predomina sobre el corazón, músculos vasculares y otros músculos lisos. Entre las acciones en sistemas de órganos están: Efectos en la presión arterial (al administrarse en forma rápida una dosis farmacológica por vía intravenosa, ocasiona un efecto característico sobre la presión arterial, que aumenta rápidamente a un máximo proporcional a la dosis), efectos vasculares (actúa principalmente sobre las arteriolas más pequeñas y los esfínteres precapilares, aunque las grandes venas y arterias también responden a ella), efectos en el corazón (actúa directamente sobre los receptores β1 predominantes en el miocardio y en las células del marcapasos y los tejidos conductores), efectos en los músculos lisos (los efectos dependen del tipo de receptor adrenérgico en el músculo. Por lo general, relaja el músculo liso GI debido a la activación de los receptores α y β), efectos respiratorios (tiene una poderosa acción broncodilatadora, más evidente cuando el músculo bronquial se contrae debido a una enfermedad, como en el asma bronquial, o en respuesta a fármacos o varios autacoides), efectos en el SNC (siendomuncompuesto polar, penetra poco en el CNS y, por tanto, no es un potente estimulante del CNS. Si bien el medicamento puede causar inquietud, aprensión, dolor de cabeza y temblores), efectos metabólicos (eleva las concentraciones de glucosa y lactato en sangre. Esta inhibe la secreción de insulina a través de una interacción con receptores α2, mientras que la activación de receptores β2 potencia las secreciones de insulina; el efecto predominante es la inhibición. La secreción de glucagón se potencia mediante la activación de los receptores β de las células α de los islotes pancreáticos), y otros efectos como la reducción del volumen de plasma circulante por la pérdida de líquido libre de proteínas hacia el espacio extracelular, aumentando así el hematócrito y la concentración de proteína plasmática. También aumenta con rapidez el número de leucocitos polimorfonucleares circulantes, probablemente debido a la demarcación mediada por el receptor β de estas células. La EPI acelera la coagulación de la sangre y estimula la fibrinólisis.
Aunque la EPI no excita directamente el músculo esquelético, facilita la transmisión neuromuscular, en particular la que sigue a la estimulación rápida prolongada de los nervios motores. La EPI también actúa directamente sobre fibras musculares blancas de contracción rápida para prolongar el estado activo, aumentando así la tensión máxima. La epinefrina promueve una caída en el plasma de K+ , en gran parte debido a la estimulación de la captación de los K+ en las células, particularmente del músculo esquelético, debido a la activación de los receptores β2. Esto se asocia con la disminución de la excreción renal de K+. La administración de dosis grandes o repetidas de EPI u otras aminas simpaticomiméticas a animales experimentales daña las paredes arteriales y el miocardio, incluso induce necrosis en el corazón indistinguible del infarto de miocardio.
La epinefrina no es efectiva tras la administración oral, porque se conjuga y se oxida rápidamente en la mucosa gastrointestinal y en el hígado. La absorción de los tejidos subcutáneos se produce de forma más o menos lenta debido a la vasoconstricción local. La absorción es más rápida después de la inyección intramuscular. Esta se inactiva rápidamente en el hígado mediante la COMT y la MAO. La EPI es inestable en solución alcalina; cuando se expone al aire o a la luz se torna rosada por oxidación hasta la forma de adrenocromo y luego se vuelve marrón por la formación de polímeros. La EPI inyectable está disponible en soluciones de 1, 0.5 y 0.1 mg/mL. Una dosis subcutánea varía de 0.3 a 0.5 mg.
La epinefrina puede causar inquietud, dolor de cabeza pulsante, temblor y palpitaciones. Los efectos disminuyen rápidamente con descanso, tranquilidad, reposo horizontal y consuelo. Las reacciones más graves incluyen hemorragia cerebral y arritmias cardiacas. El uso de dosis grandes o una inyección intravenosa rápida y accidental de EPI puede provocar una hemorragia cerebral, por el aumento brusco de la presión arterial.
¿Qué usos tiene la EPI?
Proporcionar alivio rápido y de emergencia, en caso de reacciones de hipersensibilidad, incluida la anafilaxis, a fármacos y otros alérgenos. También se usa para prolongar la acción de los anestésicos locales, presumiblemente al disminuir el flujo sanguíneo local y reducir la absorción sistémica.
- Norepinefrina
Es un importante mediador químico liberado por los nervios simpáticos posganglionares de los mamíferos. La NE constituye entre 10 y 20% del contenido de catecolaminas de la médula suprarrenal humana y hasta 97% en algunos feocromocitomas, que pueden no expresar la enzima feniletanolamina-N-metiltransferasa. Al igual que la EPI es agonista directo de las células efectoras y las acciones de estos difieren principalmente en la relación de su efectividad para estimular los receptores α y β2. Ellos son aproximadamente equipotentes en la estimulación de los receptores β1. La NE es un agonista α potente y tiene una acción relativamente pequeña sobre los receptores β2; sin embargo, es algo menos potente que la EPI en los receptores de la mayoría de los órganos. La NE tiene un potente efecto cardiovascular. En respuesta a la infusión intravenosa de NE en humanos, se incrementan las presiones sistólica y diastólica y, generalmente, la presión del pulso. El gasto cardiaco no cambia o disminuye, y aumenta la resistencia periférica total. La actividad reflejo vagal compensatoria disminuye la velocidad del corazón, superando la acción directa cardioaceleradora, y aumenta el volumen sistólico. La resistencia vascular periférica aumenta en la mayoría de los lechos vasculares y el flujo sanguíneo renal se reduce. La NE contrae los vasos mesentéricos y reduce el flujo sanguíneo esplácnico y hepático. El flujo coronario por lo general aumenta, tal vez debido tanto a la dilatación coronaria inducida indirectamente como a la EPI y la presión sanguínea elevada. El medicamento causa hiperglucemia y otros efectos metabólicos similares a los producidos por la EPI, pero éstos se observan sólo cuando se administran dosis grandes, porque la NE no es una “hormona” tan efectiva como la EPI. La inyección intradérmica de dosis adecuadas causa sudoración que no se bloquea con atropina.
La norepinefrina, como la EPI, no es efectiva cuando se administra por vía oral y se absorbe poco en los sitios de inyección subcutánea. Se inactiva rápidamente en el cuerpo por las mismas enzimas que el metilato (COMT) y deaminan por oxidación la EPI (MAO). Pequeñas cantidades se encuentran normalmente en la orina. La tasa de excreción puede aumentar mucho en pacientes con feocromocitoma. Los efectos adversos de la NE son similares a los de la EPI, aunque hay una mayor elevación típica de la presión arterial con la NE. Las dosis excesivas pueden causar hipertensión grave.
La norepinefrina se usa como un vasoconstrictor para aumentar o apoyar la presión arterial bajo ciertas condiciones de cuidados intensivos.
- Dopamina
Es el precursor metabólico inmediato de la NE y la EPI, además de ser un neurotransmisor central de particular importancia en la regulación del movimiento y posee importantes propiedades farmacológicas intrínsecas. En la periferia, se sintetiza en células epiteliales del túbulo proximal, y se cree que ejerce efectos diuréticos y natriuréticos locales. A bajas concentraciones, la interacción primaria de la DA es con los receptores vasculares D1, especialmente en los lechos renal, mesentérico y coronario. Al activar la adenilil ciclasa y elevar las concentraciones intracelulares de cAMP (adenosín monofosfato-3’,5’ cíclico), la estimulación del receptor D1 conduce a la vasodilatación. La infusión de dosis bajas de DA causa un aumento en la tasa de filtración glomerular, flujo sanguíneo renal y excreción de Na+. a. Las acciones tubulares renales de la DA que causan natriuresis se pueden ver aumentadas por el incremento en el flujo sanguíneo renal y el pequeño aumento en la tasa de filtración glomerular que sigue a su administración. El incremento resultante de la presión hidrostática en los capilares peritubulares y la reducción de la presión oncótica pueden contribuir a la disminución de la reabsorción de Na+ por las células tubulares proximales. A concentraciones más altas, la DA ejerce un efecto inotrópico positivo sobre el miocardio, actuando sobre los receptores adrenérgicos β1. La DA también causa la liberación de la NE de las terminaciones nerviosas, lo que contribuye a sus efectos sobre el corazón.
Durante la infusión de DA se pueden presentar náuseas, vómitos, taquicardia, dolor anginoso, arritmias, dolor de cabeza, hipertensión y vasoconstricción periférica. La extravasación de grandes cantidades de DA durante la infusión puede causar necrosis isquémica y esfacelo. En ocasiones excepcionales, a una infusión prolongada del fármaco ha seguido gangrena en los dedos de las manos o los pies.
Usos terapéuticos: La DA se usa en el tratamiento de la insuficiencia congestiva cardiaca grave, especialmente en pacientes con oliguria y resistencia vascular periférica baja o normal. El fármaco también puede mejorar los parámetros fisiológicos en el tratamiento del choque cardiogénico y el séptico. Si bien la DA puede mejorar de forma importante la función cardiaca y renal en pacientes gravemente enfermos con enfermedad cardiaca crónica o insuficiencia renal. . El medicamento se administra a una velocidad de 2-5 μg/kg por minuto; esta velocidad se puede aumentar gradualmente hasta 20-50 μg/kg por minuto, o más, según lo dicte la situación clínica. La duración de la acción de la DA es breve y, por tanto, la velocidad de administración puede usarse para controlar la intensidad del efecto.
Agonistas de los recetores adrenérgicos β
Este grupo de fármacos desempeñan un papel principal sólo en el tratamiento de la broncoconstricción en pacientes con asma (obstrucción reversible de las vías respiratorias) o la COPD. Los usos menores incluyen el manejo del trabajo de parto prematuro, el tratamiento del bloqueo cardíaco completo en estado de choque, el tratamiento a corto plazo de la descompensación cardíaca después de cirugía y en pacientes con insuficiencia cardíaca congestiva o infarto de miocardio. También se pueden usar para estimular la velocidad y la fuerza de la contracción cardíaca. El efecto cronotrópico es útil en el tratamiento de emergencia de arritmias tales como torsades de pointes, bradicardia o bloqueo cardiaco, mientras que el efecto inotrópico es útil cuando resulta deseable aumentar la contractilidad miocárdica.
- Isoproterenol
Es un potente agonista del receptor β no selectivo, con muy baja afinidad para los receptores α. En consecuencia, tiene efectos potentes en todos los receptores β y casi ninguna acción en los receptores α. Los efectos cardíacos pueden provocar palpitaciones, taquicardia sinusal y arritmias más graves; grandes dosis de INE causan necrosis miocárdica en animales de experimentación. El isoproterenol relaja casi todas las variedades de músculos lisos cuando el tono es alto, una acción que es más intensa en los músculos bronquial y GI lisos. El INE previene o alivia la broncoconstricción. Su efecto en el asma puede deberse, en parte, a una acción adicional para inhibir la liberación inducida por antígenos de histamina y otros mediadores de la inflamación, una acción compartida por los estimulantes selectivos de β2.
El isoproterenol se absorbe fácilmente cuando se administra por vía parenteral o en aerosol. Se metaboliza mediante COMT principalmente en el hígado, pero también en otros tejidos, no es absorbido por las neuronas simpáticas en la misma medida que la EPI y la NE. La duración de la acción del INE, por tanto, puede ser más larga que la de la EPI, pero es aún relativamente breve.
Usos terapéuticos: en casos de emergencia se utiliza para estimular la frecuencia cardiaca en pacientes con bradicardia o bloqueo cardiaco, particularmente en anticipación a la inserción de un marcapasos cardiaco artificial, o en pacientes con arritmia ventricular torsades de pointes. En trastornos como el asma y el choque, el INE ha sido reemplazado, en gran medida, por otros medicamentos simpaticomiméticos.
Efectos adversos que provoca el isoproterenol son: palpitaciones, taquicardia, dolor de cabeza y rubor son comunes. Se pueden presentar isquemia cardiaca y arritmias, particularmente en pacientes con enfermedad coronaria subyacente.
- Dobutamina
Es una amina simpaticomimética usada para el tratamiento de insuficiencia cardíaca y choque cardiogénico. Su mecanismo primario es la estimulación directa del receptor adrenérgico β1 del sistema nervioso simpático. Los efectos farmacológicos de la dobutamina se deben a las interacciones directas con los receptores α y β; sus acciones no parecen ser el resultado de la liberación de NE desde las terminaciones nerviosas simpáticas, ni son ejercidas por los receptores dopaminérgicos.
Usos terapéuticos: está indicada para el tratamiento a corto plazo de la descompensación cardíaca que puede ocurrir después de una cirugía cardíaca o en pacientes con insuficiencia cardíaca congestiva o infarto agudo de miocardio. La dobutamina aumenta el gasto cardíaco y el volumen sistólico en tales pacientes, por lo común, sin un marcado aumento en la frecuencia cardíaca.
¿Cuáles son sus efectos adversos? durante su administración, la presión arterial y la frecuencia cardiaca pueden aumentar significativamente. Los pacientes con antecedentes de hipertensión pueden mostrar una respuesta presora exagerada con mayor frecuencia. También puede aumentar el tamaño de un infarto de miocardio al incrementar la demanda de O2 del miocardio, una propiedad común de los agentes inotrópicos.
- Agonistas selectivos de los recertores adrenérgicos β2
Algunos de los principales efectos adversos de los agonistas del receptor β en el tratamiento del asma o la COPD son causados por la estimulación de los receptores β1 en el corazón. Los agentes selectivos β2 se han desarrollado para evitar estos efectos adversos. Una segunda estrategia que ha aumentado la utilidad de varios agonistas selectivos de β2 en el tratamiento del asma y la COPD ha sido una modificación estructural que da como resultado tasas más bajas de metabolismo y biodisponibilidad oral mejorada. Las modificaciones han incluido colocar los grupos hidroxilo en las posiciones 3 y 5 del anillo de fenilo o sustituir otro grupo hidroxilo en la posición 3. Una estrategia final para mejorar la activación preferencial de los receptores β2 pulmonares es la administración por inhalación de pequeñas dosis del fármaco, en forma de aerosol. Es típico que este enfoque conduzca a la activación efectiva de los receptores β2 en los bronquios, pero a concentraciones sistémicas del fármaco muy bajas.
En el tratamiento del asma y la COPD, los agonistas del receptor β se utilizan para activar los receptores pulmonares que relajan el músculo liso bronquial y disminuyen la resistencia de las vías respiratorias. Los agonistas del receptor β también pueden suprimir la liberación de leucotrienos e histamina de los mastocitos en el tejido pulmonar, mejorar la función mucociliar, disminuir la permeabilidad microvascular y, posiblemente, inhibir la fosfolipasa A2.
- Agonistas adrenérgicos β2 de acción corta
Metaproterenol es resistente a la metilación por COMT, y una fracción sustancial (40%) se absorbe en forma activa después de la administración oral. Se excreta principalmente como conjugados del ácido glucurónico. a. Los efectos ocurren minutos después de la inhalación y persisten durante varias horas. Después de la administración oral, el inicio de la acción es más lento, pero los efectos duran 3-4 h. Este fármaco se usa para el tratamiento a largo plazo de las enfermedades obstructivas de las vías respiratorias y el asma, y para el tratamiento del broncoespasmo agudo. Los efectos secundarios son similares a los de broncodilatadores simpaticomiméticos de acción corta e intermedia.
Albuterol es un agonista selectivo del receptor β2 con propiedades farmacológicas e indicaciones terapéuticas similares a las de la terbutalina. Se puede administrar por inhalación o por vía oral para el alivio sintomático del broncoespasmo. Al administrarse produce una broncodilatación significativa en 15 minutos y los efectos persisten durante 3-4 h. Los efectos cardiovasculares del albuterol son mucho más débiles que los del INE, cuando se administran por inhalación dosis que producen una broncodilatación similar. El albuterol oral tiene el potencial de retrasar el trabajo de parto prematuro. Aunque infrecuentes, a veces se observan efectos secundarios en el CNS y vías respiratorias.
Levalbuterol es el enantiómero R del albuterol, un fármaco racémico que se usa para tratar el asma y la COPD. Es β2 selectivo y actúa como otros agonistas adrenérgicos β2. En general, el levalbuterol tiene propiedades farmacocinéticas y farmacodinámicas similares a las del albuterol.
Pirbuterol es un agonista β2 relativamente selectivo. Es la única preparación disponible en un inhalador de dosis medidas activado por la respiración, un dispositivo destinado a optimizar la administración de medicamentos al liberar aerosol sólo cuando el paciente inicia la inspiración.
Terbutalina es efectivo cuando se toma por vía oral o subcutánea o por inhalación. Los efectos se observan rápidamente después de la inhalación o administración parenteral; después de la inhalación, su acción puede persistir durante 3-6 h. Con la administración oral, el inicio del efecto puede retrasarse 1-2 h. La terbutalina se usa para el tratamiento a largo plazo de las enfermedades obstructivas de las vías respiratorias y para el tratamiento del broncoespasmo agudo; también está disponible en uso parenteral para el tratamiento de emergencia en el estado asmático.
Isoetarina es el fármaco selectivo β2 más antiguo. Aunque es resistente al metabolismo por MAO, es una catecolamina y, por tanto, es un buen sustrato para COMT. En consecuencia, se usa únicamente por inhalación para el tratamiento de episodios agudos de broncoconstricción.
Fenoterol: luego de la inhalación, tiene un inicio rápido de acción y su efecto, por lo general, se mantiene durante 4-6 h. Las disritmias y los efectos cardíacos asociados con fenoterol probablemente se deben a los efectos sobre los receptores adrenérgicos β1.
Procaterol: seguido de la inhalación, tiene un rápido inicio de acción que se mantiene durante aproximadamente 5 h.
- Agonistas adrenérgicos β2 de acción prolongada (LABA)
Salmeterol: es un agonista selectivo β2 lipofílico con una duración de acción prolongada (>12 h) y una selectividad para los receptores β2 aproximadamente 50 veces mayor que la del albuterol. El salmeterol proporciona alivio sintomático y mejora la función pulmonar y la calidad de vida en pacientes con COPD. Es tan eficaz como el antagonista colinérgico ipratropio, más eficaz que la teofilina, y tiene efectos aditivos cuando se utiliza en combinación con el ipratropio inhalado o la teofilina oral. El salmeterol también puede tener actividad antiinflamatoria. El salmeterol y el formoterol son los agentes a elegir para el asma nocturna en pacientes que permanecen sintomáticos, a pesar de los agentes antiinflamatorios y otros tratamientos estándar. Por lo general, el salmeterol es bien tolerado, pero tiene el potencial de aumentar la frecuencia cardiaca y la concentración de glucosa en plasma, producir temblores y disminuir la concentración plasmática de K+ , a través de los efectos sobre los receptores β2 extrapulmonares.
Formoterol: es un agonista del receptor selectivo β2 de acción prolongada. Una broncodilatación significativa, que puede persistir hasta por 12 h, ocurre minutos después de la inhalación de una dosis terapéutica.
Arformoterol: está aprobado por la FDA para el tratamiento a largo plazo de la broncoconstricción en pacientes con COPD, que incluye bronquitis crónica y enfisema.
- Agonistas adrenérgicos β2 de acción muy prolongada (VLABA)
Indacaterol: tiene un inicio de acción rápida, aparece bien tolerado y es efectivo en la COPD con poca taquifilaxia en el uso continuo. A diferencia del salmeterol, el indacaterol no antagoniza el efecto broncorrelajante de los agonistas adrenérgicos β2 de acción corta.
Olodaterol es también un agonista β2 de acción prolongada, que se administra una vez al día, aprobado para su uso en la COPD.
Vilanterol: es un VLABA aprobado para su uso en combinación con la fluticasona. Se utiliza para mejorar los síntomas y prevenir el broncoespasmo o ataques de asma.
- Otros agonistas selectivos β2
Ritodrina: es usado como relajante uterino. Sus propiedades farmacológicas se parecen mucho a las de los otros agentes en este grupo. La ritodrina se absorbe de forma rápida, pero incompleta (30%), después de la administración oral: el medicamento puede administrarse por vía intravenosa a pacientes seleccionadas para detener el trabajo de parto prematuro.
Agonistas de los recetores adrenérgicos α
- Agonistas selectivos de los receptores adrenérgicos α1
Fenilefrina: activa los receptores β sólo a concentraciones mucho más altas. El medicamento causa una marcada vasoconstricción arterial durante la infusión intravenosa. La fenilefrina también se usa como descongestivo nasal y como midriático en diversas formulaciones nasales y oftálmicas.
Metaraminol: ejerce efectos directos sobre los receptores adrenérgicos α vasculares y actúa indirectamente al estimular la liberación de NE. El fármaco es utilizado en el tratamiento de estados hipotensivos o para aliviar, con un uso fuera de etiqueta, los ataques de taquicardia auricular paroxística, en particular los relacionados con hipotensión.
Midodrina: es un profármaco, convertido a un metabolito activo, desglumidodrina, que alcanza concentraciones máximas aproximadamente 1 h después de una dosis de midodrina. Los aumentos en la presión sanguínea inducidos por la midodrina están asociados con la contracción del músculo liso arterial y venoso.
- Agonistas selectivos de los receptores adrenérgicos α2
Se usan principalmente para el tratamiento de la hipertensión sistémica. La clonidina, un agonista α2 que se desarrolló como un vasoconstrictor descongestivo nasal, disminuye la presión sanguínea activando los receptores α2 en el CNS, suprimiendo así el flujo simpático del cerebro. Los agonistas α2 también reducen la presión intraocular, al disminuir la producción de humor acuoso. Dos derivados de la clonidina, la apraclonidina y la brimonidina, aplicados en el ojo tópicamente, disminuyen la presión intraocular, con poco o ningún efecto sobre la presión arterial sistémica.
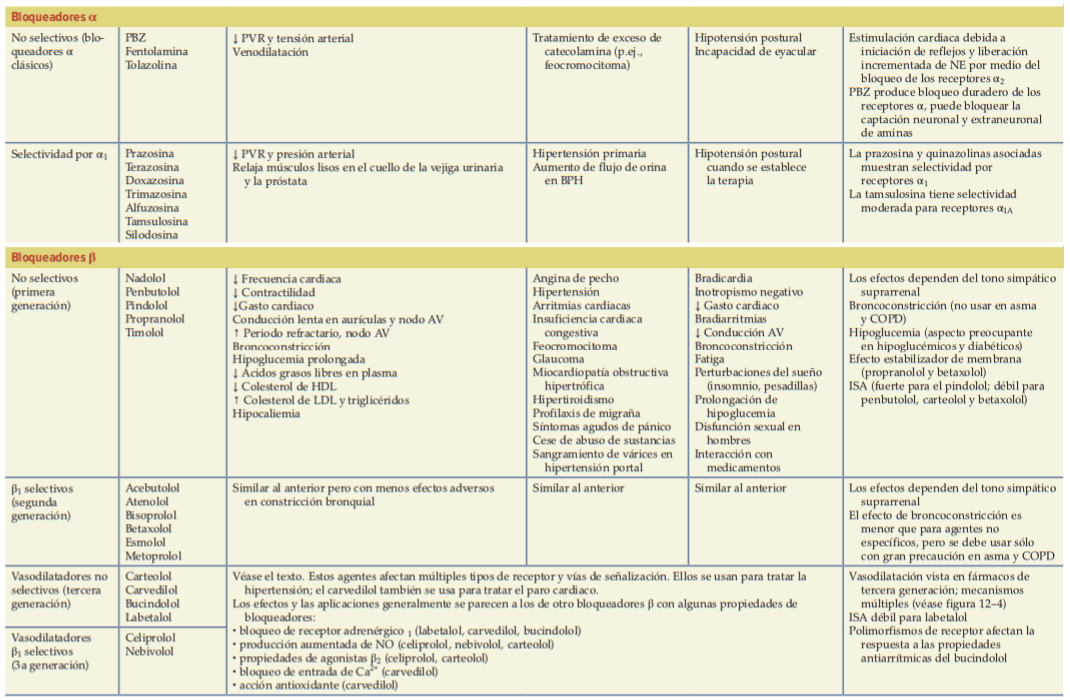
A continuación se presenta un cuadro resumen: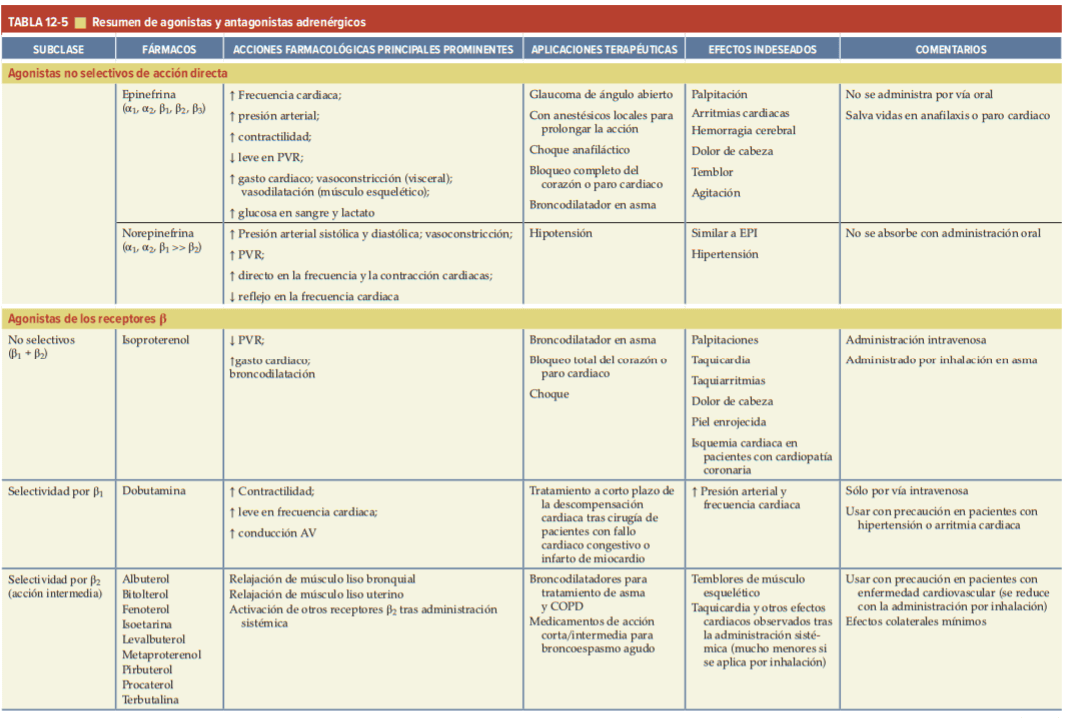
ANTAGONISTAS DE LOS RECEPTORES ADRENÉRGICOS
Un antagonista adrenérgico o bloqueante adrenérgico es una sustancia que actúa inhibiendo la acción de los receptores adrenérgicos, por lo que es un tipo de simpaticolítico y tiene acciones contrarias a los agonistas adrenérgicos.
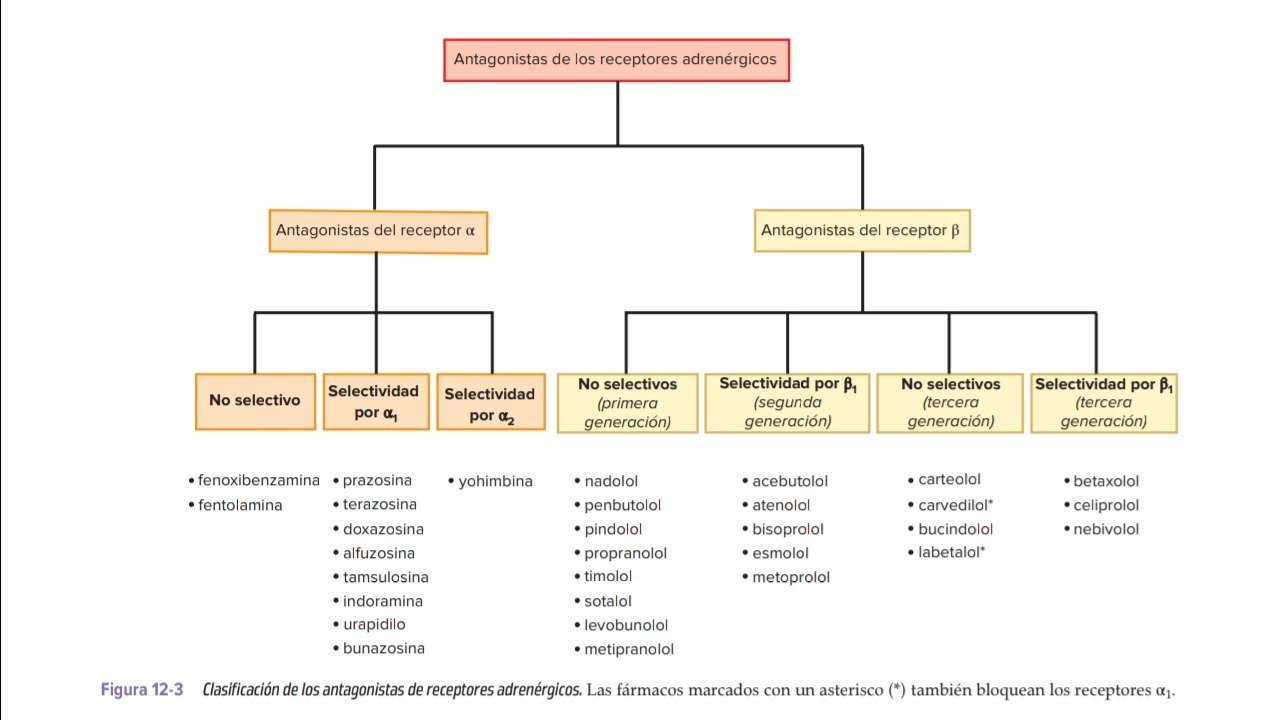
Antagonistas de los receptores adrenérgicos α
Algunos de los efectos clínicos más importantes observados de los antagonistas del receptor α están en el sistema cardiovascular. Las acciones tanto en el CNS como en la periferia están involucradas; el resultado depende del estado cardiovascular del paciente en el momento de la administración del fármaco y de la selectividad relativa del agente para los receptores α1 y α2. Los antagonistas del receptor α tienen un amplio espectro de especificidades farmacológicas y son químicamente heterogéneos.
- Antagonistas de los receptores adrenérgicos α1
Inhibe la vasoconstricción inducida por las catecolaminas endógenas; la vasodilatación puede ocurrir en ambos: vasos y venas de resistencia arteriolar. El resultado es una caída en la presión sanguínea debido a la disminución de la resistencia periférica. La magnitud de tales efectos depende de la actividad del sistema nervioso simpático en el momento en que se administra el antagonista y, por tanto, es menor en sujetos en decúbito supino que en los que están erectos, y es particularmente notable si hay hipovolemia.
Entre los agentes dispomibles se encuentran: Prazosina, Terazosina, Doxazosina, Alfuzosina, Tamsulosina y Silodosina. Estos son usados para tratar la hipertensión, insuficiencia cardíaca congestiva, entre otros.
- Antagonistas de los receptores adrenérgicos α2
La activación de los receptores α2 presinápticos inhibe la liberación de la NE y otros cotransmisores de las terminaciones nerviosas simpáticas periféricas. La activación de los receptores α2 en la región pontomedular del CNS inhibe la actividad del sistema nervioso simpático y conduce a una disminución de la presión sanguínea; estos receptores son un sitio de acción para medicamentos como la clonidina. El bloqueo de los receptores α2 con antagonistas selectivos como la yohimbina puede aumentar la corriente simpática de salida y potenciar la liberación de la NE de las terminaciones nerviosas, lo que lleva a la activación de los receptores α1 y β1 en el corazón y la vasculatura periférica, con el consiguiente aumento de la presión sanguínea.
- Antagonistas no selectivos de los receptores adrenérgicos α
Lo constituyen la Fenoxibenzamida y fentolamina.
Antagonistas de los receptores adrenérgicos β
Los antagonistas competitivos de los receptores adrenérgicos β, o bloqueadores β, han recibido una atención clínica enorme debido a su eficacia en el tratamiento de la hipertensión, la cardiopatía isquémica, la insuficiencia cardiaca congestiva y ciertas arritmias. La infinidad de antagonistas β se puede distinguir por las siguientes propiedades:
- Afinidad relativa por los receptores β1 y β2.
- Actividad simpaticomimética intrínseca.
- Bloqueo de receptores α.
- Diferencias en la solubilidad de los lípidos (penetración del CNS).
- Capacidad para inducir vasodilatación.
- Parámetros farmacocinéticos.
El propranolol es un antagonista competitivo de los receptores β y sigue siendo el prototipo con el que se comparan otros antagonistas β.
Propiedades farmacológicas
Las propiedades farmacológicas de los antagonistas de los receptores β pueden deducirse, y explicarse en gran parte, a partir del conocimiento de las respuestas provocadas por los receptores en los diversos tejidos y la actividad de los nervios simpáticos que inervan estos tejidos.
A continuación se presenta un cuadro resumen sobre antagonistas de los receptores adrenérgicos: